Computational tools for the alignment and superposition of macromolecular structures are essential instruments in structural biology. TopMatch-web provides an easy-to-use interface to a suite of techniques for protein structure alignments called TopMatch (Sippl & Wiederstein, 2008; Sippl & Wiederstein, 2012; Wiederstein & Sippl, 2020).
Given a macromolecular structure (protein or nucleic acid, =the query), the TopMatch search engine returns a list of known protein structures (=the targets) sorted by structural similarity to the query. This list is presented in a table, with various quantities describing the extent of structure similarity between query and targets. Table rows link to the respective pairwise structure comparisons.
Input
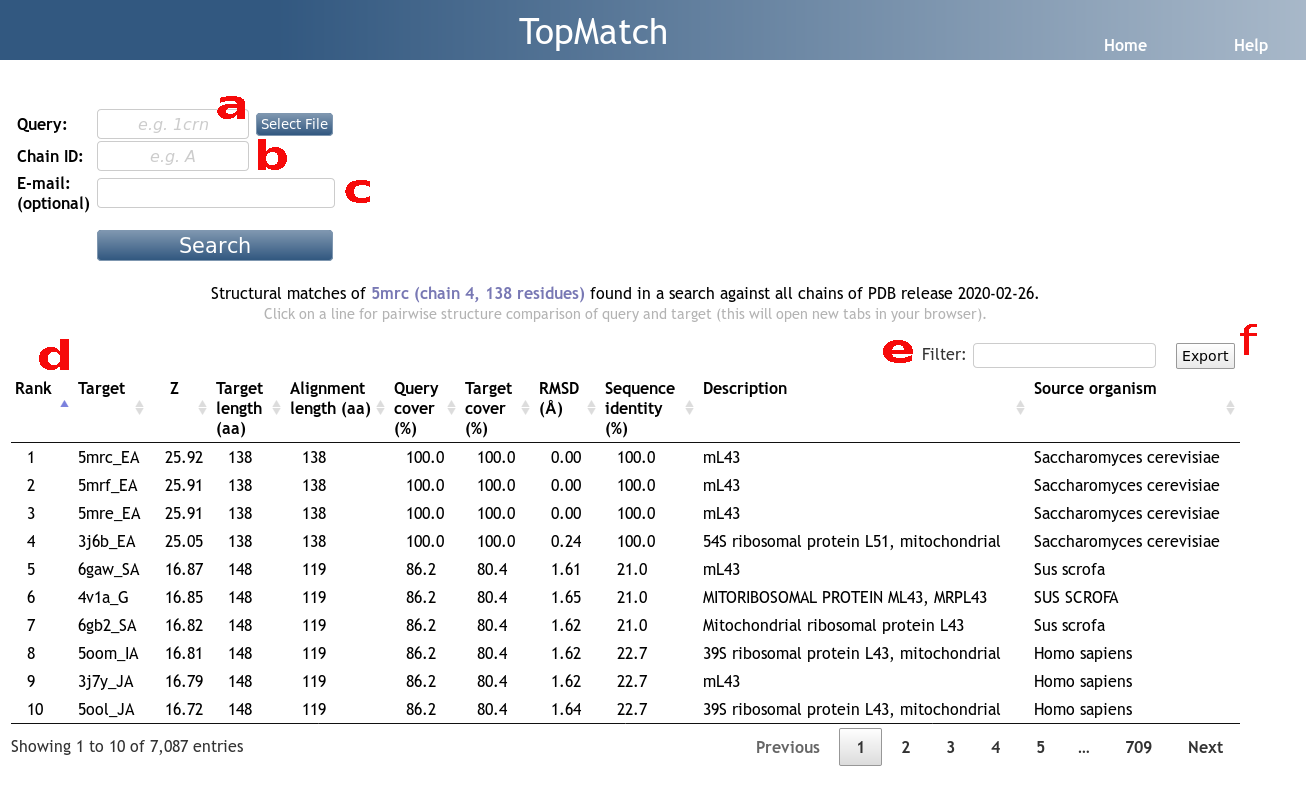
Query structure
TopMatch-web requires the atomic coordinates of the structure that is to be compared against all target structures. Users can supply coordinates either by uploading files in mmCIF format or by entering a 4-letter PDB code that refers to protein and/or nucleic acid structure available from the latest version of PDB (Figure 1a).
Furthermore, the user has to enter a chain identifier that refers to a protein/nucleic acid chain of the query (Figure 1b). Here, we use author-provided chain identifiers (as, for example, shown for PDB entries on the RCSB PDB web pages).
Further information on mmCIF format can be found in the Beginner’s Guide to PDB Structures and the PDBx/mmCIF Format provided by RCSB PDB. For conversion of files in legacy PDB format to mmCIF, the mmCIc conversion service provided by PDBj may be of help.
E-mail (optional)
If an e-mail address is given, a notification is sent to the specified address as soon as the results of the search are available (Figure 1c). In addition, a URL is displayed after the search has been started. This URL can be used to get back later and access the results.
Output
As soon as the input has been submitted, the user is informed about the job status ("queue", "processing") and provided with an URL that can be used to check for the results later. TopMatch compares the query with all chains available from the current protein data bank (PDB). Generally, search time is in the order of minutes, with variations depending on query size and server load.
Target table
TopMatch reports a list of protein or nucleic acid chains ranked by their similarity to the query structure (Figure 1d). Here, structure similarity is quantified by a score that combines the number of aligned residues derived from the respective structure alignment with the distance between each two paired residues (see Help page for pairwise TopMatch). Each alignment to a target is characterized by a small set of parameters:
- Target
-
4-letter PDB code and chain id (PDB-provided
=
label_asym_id) of target structure. - Z
-
In general, only a small fraction of the complete list
of targets will show considerable structure similarity
to the query. A simple z-score statistics is
used to keep the number of reported targets in a
reasonable range: Let S be the similarity score
calculated from the structure alignment of query and a
particular target, Smean be the mean
score over all targets, and Ssdev be
the corresponding standard deviation.
Then z = (S-Smean)/Ssdev. TopMatch-web only reports targets with a z-score ≥ 2.9. - Target length
- Number of residues in the target structure.
- Alignment length
- Number of residue pairs that are structurally equivalent (subsequently referred to as LEN).
- Query cover
-
Query cover based on alignment length, expressed in
percent
(= 100 x LEN/Qn, where Qn is the number of residues in the query structure). - Target cover
-
Target cover based on alignment length, expressed in
percent
(= 100 x LEN/Tn, where Tn is the number of residues in the target structure). - RMSD
- Root-mean-square error of superposition in Ångström, calculated using all structurally equivalent C-alpha (proteins) or P (nucleic acids) atoms.
- Sequence identity
- Sequence identity of query and target in the equivalent regions, expressed in percent.
- Description
- A short description of the function/molecule name of the target chain.
- Source organism
- The scientific name of the organism which the target chain was derived from.
- Ligands
- Three-letter codes of all ligands found in the target entry, separated by '//'.
To examine the target table more efficiently, it is possible to sort the list by clicking on the respective column headers. By clicking on the header again, the current mode of sorting is reversed.
Sometimes it might be convenient to be able to search for specific PDB codes, organisms, etc. in the list of targets. Entering a search term into the box labeled "Filter" restricts the results to entries that contain the specified term (Figure 1e).
The table can be downloaded in CSV format by clicking on the "Export" button (Figure 1f).
A particular target can be selected by clicking on the respective line in the target table. This will open a new tab in the web browser showing the pairwise structure comparison of the query and the selected target.
System Requirements
TopMatch-web should run on most operating systems with all common browsers. We have successfully used TopMatch-web in the following environments:
| OS | Version | Chrome | Firefox | Safari | Microsoft Edge | Opera |
|---|---|---|---|---|---|---|
| Linux | Debian 8 | 57.0 | 60.4 | n/a | n/a | not tested |
| Linux | Ubuntu 18.04 | not tested | 64.0 | n/a | n/a | not tested |
| MacOS | 10.12.6 | not tested | 62.0 | 12.0 | n/a | not tested |
| MacOS | 10.9.5 | 65.0 | not tested | 9.1.3 (not working) | n/a | 49.0 |
| Windows | 10 | 71.0 | 64.0 | n/a | 42.17134 | not tested |
If you experience any problems using TopMatch, please feel free to contact us.
References
Wiederstein & Sippl (2020) TopMatch-web: pairwise matching of large assemblies of protein and nucleic acid chains in 3D. Nucleic Acids Research, Web Server Issue 2020. [view]
Sippl & Wiederstein (2012) Detection of correlations in protein structures and molecular complexes. Structure 20, 718-728. [view]
Slater, Castellanos, Sippl & Melo (2012) Towards the development of standardized methods for comparison, ranking and evaluation of structure alignments. Bioinformatics 29, 47-53. [view]
Sippl & Wiederstein (2008) A note on difficult structure alignment problems. Bioinformatics 24, 426-427. [view]
Sippl (2008) On distance and similarity in fold space. Bioinformatics 24, 872-873. [view]