Computational tools for the alignment and superposition of macromolecular structures are essential instruments in structural biology. TopMatch-web provides an easy-to-use interface to a suite of techniques for macromolecular structure alignments called TopMatch (Sippl & Wiederstein, 2008; Sippl & Wiederstein, 2012; Wiederstein & Sippl, 2020).
Given a pair of structures, TopMatch calculates a list of alignments ordered by structural similarity. The best alignments are reported in a table. The corresponding superpositions can be explored in a 3D molecule viewer which highlights the structurally equivalent parts of the macromolecules. The sequence alignments resulting from the structure comparison are provided on-line and in PDF format. Coordinates of the input structures after superposition are available for download.
Input
Structures
TopMatch-web requires the atomic coordinates of the two structures that are to be compared. We term the first structure query and the second structure target. Users can supply coordinates either by uploading files in mmCIF format or by entering strings that refer to protein and/or nucleic acid structures available from the latest version of PDB. The syntax of this string allows quick access to all PDB structures and is defined as follows:
- <4-letter code>
-
Refers to the entirety of all protein and nucleic acid chains contained in a
PDB file.
- 1aqk
Examples: - <4-letter code>,<chain ID>(,<chain ID>...)
-
Refers to one or more protein/nucleic acid chain(s) of a PDB file, specified by
its 4-letter code and one ore more chain identifiers (author-provided).
- 1aqk,L
- 1aqk,L,H
Examples: - <4-letter code>_<chain ID>(_<chain ID>...)
-
Refers to one or more protein/nucleic acid chain(s) of a PDB file, specified by
its 4-letter code and one ore more chain identifiers (PDB-provided).
- 1aqk_A
- 1aqk_A_B
Examples: - <4-letter code>@<ID of biological assembly>
-
Refers to a specific assembly thought to be a
functional form of the macromolecules involved (also sometimes referred to as
the
biological unit; usually referred to by a number).
- 3f1w@2
Examples: -
<4-letter code>;<model number>
<4-letter code>_<chain ID>;<model number>
<4-letter code>,<chain ID>;<model number>
-
For structures determined by NMR a model number can be specified. If omitted, TopMatch-web uses the first model
found in the given PDB entries.
- 1kdx,A;10
- 1kdx;10
Examples:
Links to examples of various size and complexity are also available from the TopMatch-web start page.
Further information on mmCIF format can be found in the Beginner’s Guide to PDB Structures and the PDBx/mmCIF Format provided by RCSB PDB. For conversion of files in legacy PDB format to mmCIF, the mmCIF conversion service provided by PDBj may be of help.
Output
Superposition
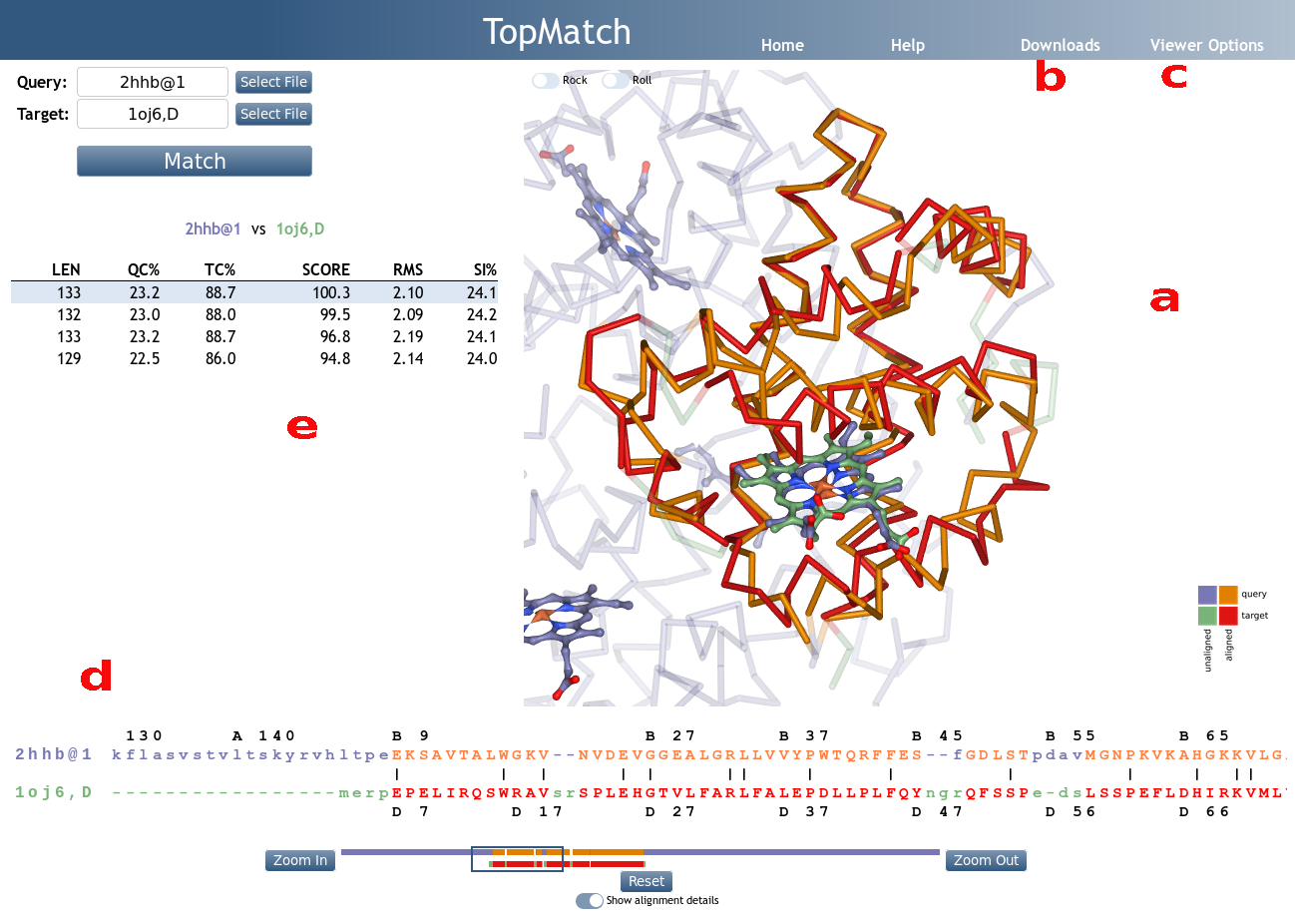
TopMatch-web visualizes the superimposed input
structures in 3D using the interactive molecule viewer
NGL
(Figure 1a). The query structure is shown in blue, the
target structure is shown in green. Pairs
of residues that are structurally equivalent are colored orange (query) or red
(target).
Transformed atomic coordinates for query and target after
superposition can be downloaded in mmCIF format or as a PyMOL script,
which also includes commands for coloring the equivalent parts of the
structures. Furthermore, the transformation matrix used to
transform the target coordinates is available in plain text format
(Figure 1b).
The representation mode of query and target can be
controlled via the "Viewer Options" menu (Figure 1c). From
there, query and target can also be selectively hidden
to facilitate the analysis. Further options include the
display of ligands, the background color, the opacity of
unmatched parts, and the size of the NGL widget.
Aligned sequences
The sequence alignment that results from a selected
structure alignment is shown in a box below the NGL widget
(Figure 1d). By default, the alignment is displayed
schematically; it can be viewed in more detail by toggling
the "Show alignment details" button. In accordance with the
3D visualization, the query sequence is colored blue, the target sequence is colored green and pairs of structurally equivalent residues are colored orange (query) or red (target).
We emphasize that in structure based sequence alignments of
this kind only the orange/red parts show meaningful
information and that the positions of the blue and green
residues are of no relevance. Two rulers showing the PDB
chain IDs and residue numbers found in the input structures
facilitate navigation.
The alignment is also typeset using the TeXshade package
(Beitz, 2000) and provided as PDF file and in FASTA format
(Figure 1b). In the PDF file, structurally equivalent
residues are marked by bars in orange (query) or red (target) and are shown in bold if they are the same amino
acid/nucleotide. The FASTA file is plain text;
structurally equivalent residues are shown as CAPITAL
letters, and blanks indicate permuted blocks.
Table of alternative alignments
TopMatch reports a ranked list of alignments (Figure 1e). The alignments are characterized by a small set of parameters:
- LEN
-
Number of residue pairs that are structurally equivalent
(= alignment length). - QC%
-
Query cover based on alignment length, expressed in
percent
(= 100 x LEN/Qn, where Qn is the number of residues in the query structure). - TC%
-
Target cover based on alignment length, expressed in
percent
(= 100 x LEN/Tn, where Tn is the number of residues in the target structure). - SCORE
- Measure of structural similarity (see Wiederstein & Sippl (2020)). If the structurally equivalent parts in query and target match perfectly, S is equal to L. With increasing spatial deviation of the aligned residues, S approaches 0.
- RMS
- Root-mean-square error of superposition in Ångström, calculated using all structurally equivalent C-alpha (proteins) or P (nucleic acids) atoms.
- SI%
- Sequence identity of query and target in the equivalent regions, expressed in percent.
A particular alignment can be selected by clicking on the respective line in the alignment table. The superposition corresponding to the selected alignment is then shown in the NGL widget.
The alignment table can be exported in CSV format.
System Requirements
TopMatch-web should run on most operating systems with all common browsers. We have successfully used TopMatch-web in the following environments:
| OS | Version | Chrome | Firefox | Safari | Microsoft Edge | Opera |
|---|---|---|---|---|---|---|
| Linux | Debian 8 | 57.0 | 60.4 | n/a | n/a | not tested |
| Linux | Ubuntu 18.04 | not tested | 64.0 | n/a | n/a | not tested |
| MacOS | 10.12.6 | not tested | 62.0 | 12.0 | n/a | not tested |
| MacOS | 10.9.5 | 65.0 | not tested | 9.1.3 (not working) | n/a | 49.0 |
| Windows | 10 | 71.0 | 64.0 | n/a | 42.17134 | not tested |
If you experience any problems using TopMatch, please feel free to contact us.
References
Wiederstein & Sippl (2020) TopMatch-web: pairwise matching of large assemblies of protein and nucleic acid chains in 3D. Nucleic Acids Research, Web Server Issue 2020. [view]
Sippl & Wiederstein (2012) Detection of correlations in protein structures and molecular complexes. Structure 20, 718-728. [view]
Slater, Castellanos, Sippl & Melo (2013) Towards the development of standardized methods for comparison, ranking and evaluation of structure alignments. Bioinformatics 29, 47-53. [view]
Sippl & Wiederstein (2008) A note on difficult structure alignment problems. Bioinformatics 24, 426-427. [view]
Sippl (2008) On distance and similarity in fold space. Bioinformatics 24, 872-873. [view]
Rose, Bradley, Valasatava, Duarte, Prlić & Rose (2018) NGL viewer: web-based molecular graphics for large complexes. Bioinformatics 34, 3755-3758. [view]
Beitz (2000) TEXshade: shading and labeling of multiple sequence alignments using LATEX2 epsilon. Bioinformatics 16, 135-139. [view]